Actu+ 7 : Comment concevoir un dispositif médical ?
Les DM et DMA :
Ce sont les dispositifs d’usage individuel (aides techniques, matériel dentaire, matériel
ophtalmologique et optique, matériel à usage unique ou réutilisable, textiles techniques, etc) ou d’équipement (appareils médicaux électroniques à visée thérapeutique, dispositifs
anesthésiques et respiratoires, matériel d’hôpital, etc). Un DM Actif tire l’énergie nécessaire à
son fonctionnement d’une source externe (batterie, secteur, air comprimé, …)
Les DMI et DMIA :
Ce sont les dispositifs implantables, actifs ou non. Ils sont prévus pour fonctionner dans le
corps humain définitivement où sur une longue durée. Parmi ces DM, on compte les prothèses
de hanche (DMI), les stimulateurs cardiaques (DMIA), …
Les DMDIV :
Ce sont les dispositifs permettant d’analyser ou d’examiner In Vitro des échantillons provenant
du corps humain. Ils participent à l’établissement d’un diagnostic. Parmi ces DMDIV, on compte
les tests de dépistage (grossesse, COVID, VIH, …) ainsi que les machines de laboratoire
d’analyse médicale.
Les dispositifs médicaux sont régis en Europe par les règlements (UE)2017/745 pour les DMA et
DMIA, et (UE)217/746 pour les DMDIV. Aux Etats-Unis, c’est la FDA qui régule les dispositifs
médicaux avec la 21 CFR 860. Dans ce document, le focus est mis sur les procédures européennes.
En plus des catégories, une classe de risque est attribuée aux DM :
- La Classe I : Pour les dispositifs à faible risque (orthèses, seringues, lunettes, fauteuil roulant, …). On ajoute à cette classe 3 sous-classes qui sont Is pour les dispositifs délivrés stériles, Im pour les dispositifs de mesurage (thermomètres, tensiomètres, …) et Ir pour les dispositifs chirurgicaux réutilisables (scalpels, pinces, …).
- La Classe IIa : Pour les dispositifs à risque potentiel modéré, en contact de courte durée (~<30j) avec le patient.
- La Classe IIb : Pour les dispositifs à risque potentiel important, en contact prolongé avec le patient.
- La Classe III : Pour les dispositifs à risque important, implanté à demeure, destinés à soutenir la vie (défibrillateurs automatiques).

C’est au fabricant d’un dispositif médical de déterminer la classe de son produit en s’appuyant sur les règles de classification définies par les directives européennes (directives DM), et en fonction de la finalité médicale revendiquée par le produit. L’annexe VIII du règlement MDR indique 22 règles pour définir la classe d’un DM, soit près de 80 critères à évaluer.
Démonstration de la conformité
Un dispositif médical, pour être mis sur le marché européen, doit être conforme aux différentes
directives en vigueur le concernant et avoir obtenu son marquage CE.
Pour les dispositifs traités par MAATEL (les DM actifs électronique), les directives essentielles
sont généralement :
- Le Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux
- Le Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro
- La directive Machines : Règlement (UE) 2023/1230 du Parlement européen et du Conseil du 14 juin 2023 sur les machines.
- La directive CEM : Directive 2014/30/UE du Parlement européen et du Conseil du 26 février 2014 relative à l’harmonisation des législations des États membres concernant la compatibilité électromagnétique.
- La directive DBT : Directive 2014/35/UE du Parlement européen et du Conseil du 26 février 2014 relative à l’harmonisation des législations des États membres concernant la mise à disposition sur le marché du matériel électrique destiné à être employé dans certaines limites de tension.
- La directive RoHS : Directive 2011/65/UE du Parlement européen et du Conseil du 8 juin 2011 relative à la limitation de l’utilisation de certaines substances dangereuses dans les équipements électriques et électroniques
La démonstration que le dispositif satisfait aux exigences des normes harmonisées associées aux
règlements, permet d’affirmer qu’il est conforme à la législation européenne en vigueur
(présomption de conformité). Les principales normes harmonisées qui concernent le dispositif à développer sont, pour les aspects médicaux :
- ISO 13485 : Dispositifs médicaux — Systèmes de management de la qualité,
- EN 60601 : Exigences générales pour la sécurité de base et les performances essentielles des DEM (Dispositifs Electro-Médicaux),
- ISO 14971 : Application de la gestion des risques aux dispositifs médicaux.
Pour les autres directives, on pourra se référer aux normes habituelles du secteur de
l’électronique.
Seules les DM de Classe I sont auto-certifiables par le fabricant. Pour toutes les autres classes,
l’intervention d’un organisme notifié est nécessaire à l’obtention de la certification ou marquage
CE.
Pour compléter la démonstration, le fabricant doit réaliser une évaluation clinique (évaluation
des publications scientifiques pertinentes, des résultats de toutes les investigations / essais
cliniques disponibles) ; voire des investigations / essais cliniques pour les DM implantables ou de
Classe III.
Il est également demandé que le fabricant mette en place un plan de surveillance après
commercialisation (cf Annexe III du règlement (UE)2017/745).
Gestion des risques
Si un dispositif médical se doit d’être efficace dans sa fonction de soin, il est également crucial
qu’il garantisse la sécurité du patient ainsi que du personnel de santé amené à le manipuler.
La gestion des risques est donc une activité fondamentale dans le développement d’un dispositif
médical. Elle consiste à identifier les dangers et situations dangereuses, à évaluer les risques,
imaginer des mesures de réduction de ces risques et mesurer leur efficacité.
Pour cela, l’ISO 14971 spécifie la terminologie, les principes et un processus de gestion des risques
relatifs aux dispositifs médicaux.
L’ISO 14971 impose aux fabricants d’établir des critères objectifs d’acceptabilité des risques, mais
ne spécifie pas de niveaux de risque acceptables.
« Le fabricant réduit les risques et traite des aspects relatifs à la sécurité d’un dispositif médical, y
compris l’acceptabilité des risques résiduels. Le fabricant tient compte de l’état de l’art
généralement admis, afin de déterminer si un dispositif médical est apte à être mis sur le marché
pour son utilisation prévue. »
Pour compléter l’approche qualitative de l’ISO 14971, il est possible de se raccrocher à d’autres
normes de sécurité fonctionnelle telles que :
- ISO 13849 : sécurité des machines,
- IEC 62061 : sécurité fonctionnelle des machines,
- EN 61508 : la sécurité fonctionnelle des systèmes électriques/électroniques/électroniques programmables (E/E/EP) relatifs à la sécurité,
- ISO 10218 : Robots et dispositifs robotiques.
Une norme importante dans le domaine de la sécurité du logiciel des dispositifs médicaux, est
aussi à prendre en considération : c’est l’IEC 62304 (Logiciels de dispositifs médicaux — Processus du cycle de vie du logiciel). Elle définit des classes (A, B, C) de sécurité pour les logiciels des DM et fournit les exigences applicables à chaque tâche du cycle de développement du logiciel.
La gestion des risques s’opère tout au long du processus de développement du DM. Ce qui vaut également pour les fonctions purement médicales du DM (innocuité du traitement, recherche des effets secondaires, biocompatibilité des matériaux, …
En tant que sous-traitant, MAATEL n’est pas en charge de l’analyse de risque. Celle-ci doit être
menée par le fabricant du DM. Par contre, nous pouvons y contribuer activement, en particulier
pour les aspects de réduction des risques, dans la limite de notre périmètre.
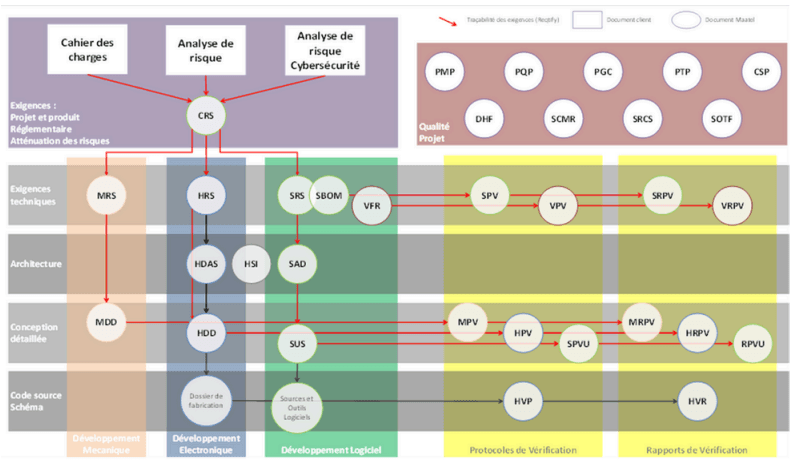
Processus de développement
MAATEL a mis la gestion du risque au cœur de son processus
de développement grâce à l’administration rigoureuse des
données d’entrée et de sortie à chaque étape, à une
architecture documentaire pertinente et précise et la
planification des ressources.